Topics
>RESEARCH
Insights into Redox-Independent Cellular Stress Response
Researchers identify the regulatory pathway that governs a type of redox-independent stress response involving NRF2
Nuclear factor erythroid 2-related factor 2 (NRF2) is a master transcription factor with a role in regulating oxidative stress response. It can be activated via redox-dependent and independent pathways. Regulatory mechanisms of the redox-dependent pathway are known; however, the same cannot be said for the redox-independent pathway. Now, researchers from Juntendo University have unraveled the roles of phase-separated liquid droplets, p62 bodies, in redox-independent NRF2 activation and the significance of phosphorylation in this process.
Researchers in Japan unravel the mechanisms of redox-independent NRF2 stress response
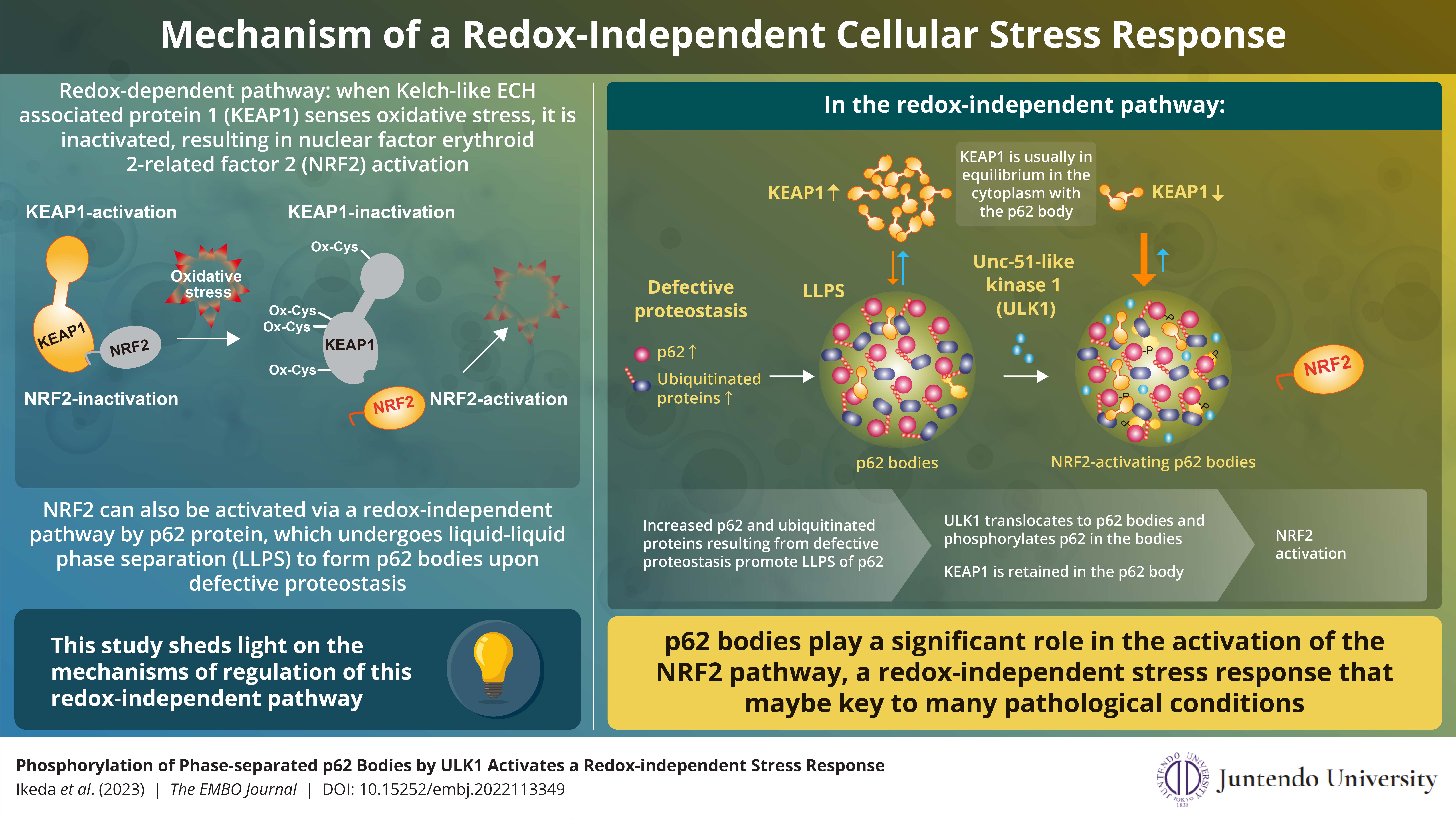 ULK1 interacts directly with and phosphorylates the biomolecular condensate, p62 body. Phosphorylated p62 retains KEAP1 within itself and activates NRF2. The findings shed new light on the redox-independent activation of NRF2, which has an important role
ULK1 interacts directly with and phosphorylates the biomolecular condensate, p62 body. Phosphorylated p62 retains KEAP1 within itself and activates NRF2. The findings shed new light on the redox-independent activation of NRF2, which has an important role
in stress response.
Image credits:Masaaki Komatsu from Juntendo University School of Medicine
License type:Original Content
Usage restrictions: You are free to share and adapt the Infographic material but attribution is required, with a link to the news source.
Cellular stress, or oxidative stress, occurs when there is a buildup of reactive oxygen species (ROS), which interferes with cellular mechanisms and can even cause damage to proteins, lipids, and DNA. Owing to their destructive nature, all cells have
robust mechanisms in place to remove ROS and reduce oxidative stress. One such mechanism is the nuclear factor erythroid 2-related factor 2 (NRF2)-mediated stress response, where NRF2 is a master transcription factor that aids in reducing oxidative stress.
Much is known about the redox-dependent activation of NRF2 and its subsequent role in stress response. In this pathway, Kelch-like ECH-associated protein 1 (KEAP1) senses oxidative stress in the cell through oxidation of its specific cysteine residues.
This oxidation causes conformational changes in KEAP1, which, in turn, loses the ability to suppress NRF2. As a result, NRF2 is stabilized and induces a series of genes encoding anti-oxidative proteins that reduce and remove oxidative stress
caused by ROS.
NRF2 can also be activated in a redox-independent manner. This activation involves p62 protein, which undergoes liquid-liquid phase separation to form p62 bodies when it binds to ubiquitinated proteins upon defective proteostasis. However, the
precise mechanism of NRF2 regulation through p62 bodies had hitherto remained largely unknown.
Now, researchers in Japan have found how p62 bodies control redox-independent NRF2 activation. They conducted a study that was conceived by Professor Masaaki Komatsu and Associate Professor Yoshinobu Ichimura from Juntendo University School of Medicine
and Dr. Nobuo N. Noda from Hokkaido University. “We have reported in a previous study that phosphorylation of p62 inhibits the binding of KEAP1 to NRF2 competitively, thereby disabling the NRF2-repressive function of KEAP1. However, the regulatory mechanism and the physiological functions in vivo remain largely unclear. This is important as the accumulation of phosphorylated p62 has been found to cause many intractable diseases,” explains
Prof. Komatsu when asked about the team’s motivation for pursuing the research. Their findings are all set to be published in The EMBO Journal.
Using advanced techniques like high-speed atomic force microscopy, fluorescence recovery after photobleaching, and fluorescence loss in photobleaching, the team conducted experiments that included those performed outside a living organism (in vitro)
and those using cells and mice (in vivo) to thoroughly profile protein-protein interactions, cellular localization of specific components, and the effects of LLPS-induced p62 phosphorylation, during redox-independent NRF2 activation.
Summarizing the main findings of their study, Dr. Ichimura explains, “We found that ULK1, a protein kinase, translocates to p62 bodies and then phosphorylates p62 within the bodies. The resulting phosphorylated p62 bodies retain KEAP1 within them, which drives the activation of NRF2.”
KEAP1 usually cycles in and out of p62 bodies; however, phosphorylated p62 tightly binds to KEAP1, which causes KEAP1 to be retained and sequestered within the p62 body. This leads to activation of even more NRF2. The p62 bodies are degraded
by autophagy, and this may contribute to shutdown of this pathway. The current study extends the scope of the antioxidative stress response and provides new insights into the role of phase separation in the process.
The importance of this redox-independent NRF2 activation was tested using phosphomimetic p62 knock-in mouse models where hyperactivation of NRF2 by extensive phosphorylation resulted in hyperkeratosis—a growth defect causing the outer layer
of the stomach and esophageal lining to thicken, which in turn caused stunted growth due to malnutrition.
Prof. Komatsu is confident that his team has laid the foundation for future work to probe deeper into the mechanism and regulation of redox-independent stress responses. He concludes, “Whether redox-dependent or independent, NRF2 activation is an important biological defense system. Understanding its regulatory mechanisms is crucial as its persistent activation leads to inordinate defense responses, like excessive keratinization. Our study is the first scientific validation of the physiological significance for redox-independent NRF2 activation. In the case of redox-independent stress responses, activation of NRF2 is very likely regulated by phosphorylation, dephosphorylation, and autophagic degradation of p62 bodies. p62 bodies have been found to accumulate in affected cells in patients with liver disorders, neurodegenerative diseases, and cancers. Thus, research such as ours will be useful in elucidating the pathogenesis of and developing improved therapies for p62-,
NRF2-, and autophagy-related diseases.”
About Professor Masaaki Komatsu from Juntendo University
Dr. Masaaki Komatsu is a Professor in the Department of Organ and Cell Physiology at Juntendo University School of Medicine. He earned his Ph.D. in 2001 from the Juntendo University School of Medicine and researches autophagy, the UFM1-system, protein
homeostasis, and organelle homeostasis. He has published over 190 peer-reviewed articles since 1999.
Reference
| Authors | Ryo Ikeda1,2, Daisuke Noshiro3, Hideaki Morishita1, Shuhei Takada1, Shun Kageyama1, Yuko Fujioka3, Tomoko Funakoshi1, Satoko Komatsu-Hirota1, Ritsuko Arai4, Elena Ryzhii4, Manabu Abe5, Tomoaki Koga6, Hozumi Motohashi7, Mitsuyoshi Nakao6, Kenji Sakimura5, Arata Horii2, Satoshi Waguri4, Yoshinobu Ichimura1, Nobuo N Noda3, and Masaaki Komatsu1 |
| Title of original paper | Phosphorylation of phase-separated p62 bodies by ULK1 activates a redox-independent stress response |
| Journal | The EMBO Journal |
| DOI | 10.15252/embj.2022113349 |
| Affiliations | 1Department of Physiology, Juntendo University Graduate School of Medicine, Japan 2Department of Otolaryngology Head and Neck Surgery, Niigata University Graduate School of Medical and Dental Sciences, Japan3Institute for Genetic Medicine, Hokkaido University, Japan 4Department of Anatomy and Histology, Fukushima Medical University School of Medicine, Japan 5Department of Animal Model Development, Brain Research Institute, Niigata University, Japan 6Department of Medical Cell Biology, Institute of Molecular Embryology and Genetics, Kumamoto University, Japan 7Department of Gene Expression Regulation, Institute of Development, Aging and Cancer, Tohoku University, Japan |